The adenylosuccinate lyase (ADSL) deficiency is a rare inherited metabolic disorder that leads to severe neurological symptoms. The rate of disability varies and the
disease can be clinically classified into four basic types: neonatal
form (prenatal hyperkinesis, lung hypoplasis, and prenatal growth abortion,
following by fatal neonatal encephalopathy) [1], severe infant form – type
I (heavy psychomotor retardation, frequently early death) [2-3], moderate to
mild form – type II (psychomotor retardation, hypotony and autism) [2, 4-5] and
very mild form [6].
ADSL (EC 4.3.2.2) is an enzyme that catalyzes the conversion of succinylaminoimidazole carboxamide ribotide (SAICAR) into aminoimidazole carboxamide ribotide (AICAR) in the de novo purine
synthesis pathway, and the formation of adenosine monophosphate (AMP) from
adenylosuccinate (SAMP) in the purine nucleotide cycle [7-8].
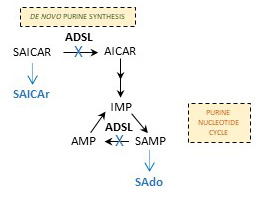
The ADSL deficiency (OMIM 103050) is an inherited autosomal recessive disorder caused by mutations in ADSL gene. The pathogenic mechanism
leading to the development of individual clinical symptoms and the underlying phenotypic heterogeneity remains unclear. The main pathogenic effect has been
attributed to the toxic effects of accumulating dephosphorylated substrates of ADSL
- SAdo and SAICAr in urine, plasma and cerebrospinal fluid [9]. Although the
absolute SAdo and SAICAr concentrations do not correlate with the severity of
the phenotype, the ratio of SAdo/SAICAr in cerebrospinal
fluid was found to correspond to the severity of the disease [10]. To date, about 109 patients with ADSL deficiency have been reported.
Biochemical diagnostics
of the disease is based on the detection of SAdo and SAICAr in urine and/or plasma, rarely in cerebrospinal fluid, and the molecular biology diagnostics on the
detection of mutations in the ADSL gene. Both are available in our laboratories. Through the GeneTests system, we provide a complex diagnostics to clinical sites not only in Czech Republic, but also abroad. In addition, we offer the commercially unavailable SAdo and SAICAr standards to other diagnostic and scientific laboratories around the world.
There is currently no cure for this rare disease, however, supportive care such as anticonvulsants is available.
[1] Mouchegh, K., et al., J Pediatr, 2007. 150(1): p. 57-61 e2.
[2] Jaeken, J., et al.,Eur J Pediatr, 1988. 148(2): p. 126-31.
[3] Maaswinkel-Mooij, P.D., et al., J Inherit Metab Dis, 1997. 20(4): p. 606-7.
[4] Jaeken, J., et al., J Inherit Metab Dis, 1992. 15(3): p. 416-8.
[5] Valik, D., P.T. Miner, and J.D. Jones, Pediatr Neurol, 1997. 16(3): p. 252-5.
[6] Mastrogiorgio, G., et al., Orphanet J Rare Dis, 2021. 16(1): p. 112.
[7] Ciardo, F., C. Salerno, and P. Curatolo, J Child Neurol, 2001. 16(5): p. 301-8.
[8] Jaeken, J. and G. Van den Berghe, 1984. 2(8411): p. 1058-61.
[9] Stone, T.W., et al., Adv Exp Med Biol, 1998. 431: p. 185-9.
[10] Van den Bergh, F., et al., J Inherit Metab Dis, 1993. 16(2): p. 415-24.